Pathoscope
The Pathoscope workflow is used for detecting known reference OTUs in an Illumina sample library.
How Does it Work?
Sample reads are mapped against a Virtool reference and reads matching OTUs are counted and used to arrive at a detection result. To understand this section, you must be familiar with references and OTUs.
The first step of the workflow involves mapping sample reads against default isolates of all OTUs in the target reference. This can be thought of as a fishing or prescreening step for OTUs that may be potentially represented in the sample. Any OTUs with at least one matching read are retained as candidate hits for the second step.
The second step begins with the creation of a Bowtie2 mapping index containing all isolates of the OTUs identified in the default isolate mapping. This step provides a more diverse mapping target for each virus and allows isolate-level detection.
A major complication of this diagnostic method is multi-mapping, the situation in which single reads find multiple mapping positions in a set of reference sequences. We can make the assumption that there are only a small number of real source sequences in the sample. In practice, reads with a single real source sequence find multiple mapping locations in Virtool references. This is especially true when OTUs contain large numbers of similar isolates.
Virtool uses the Pathoscope2 library to reassign multi-mapped reads to their most likely source sequences. An expectation maximization (EM) algorithm is used to perform the reassignment. Pathoscope makes the assumption that source sequences with high numbers of uniquely mapped (not multi-mapped) reads are most likely to be real source sequences. Read counts are reassigned fractionally when it is assigned to multiple genomes after reassignment.
Quality
Analysis quality is determined by running Pathoscope and/or NuVs on your sample of interest. These analyses can be found under the Analyses tab.
Click on the PathoscopeBowtie.
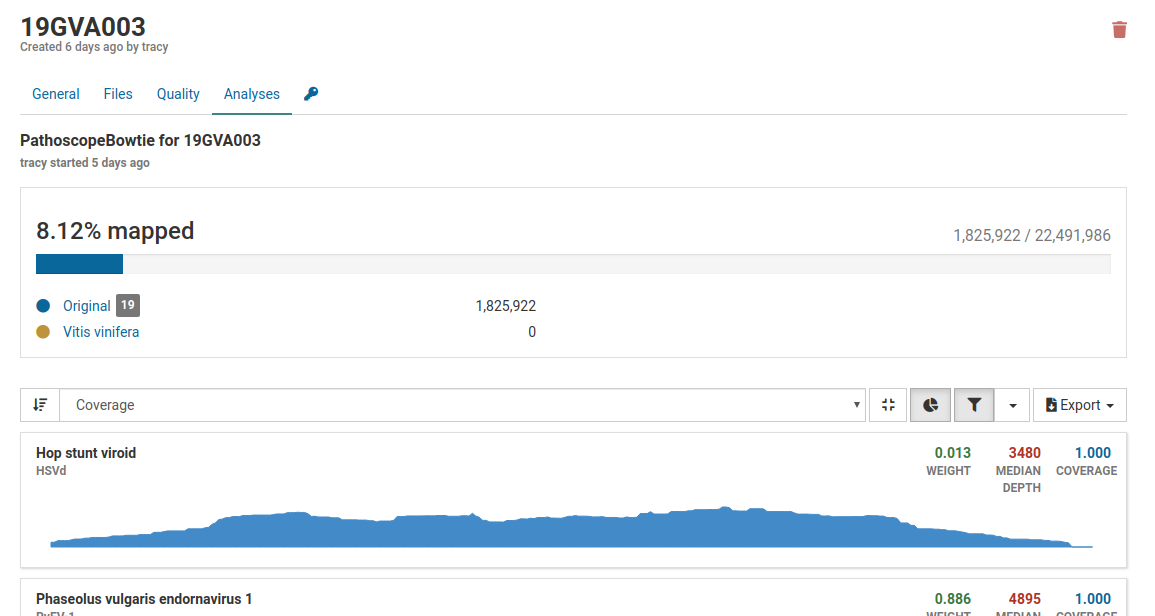
Pathoscope is the primary tool in Virtool used for determining whether a known virus is present in a sample. A number of statistics are used to determine the presence of a pathogen.
Generally, approximately 5 million reads is a good base line for a dsRNA library, however the percentage of mapped reads is of greater importance. For dsRNA, percentages can range from less than 1% to greater than 80%. The greater the enrichment of viral RNA (i.e. the higher the percent of mapped reads) the less number of total reads are required. For example, 2% of 5 million reads or 100 000 mapped reads is good.
Additional statistics include:
Coverage: a measure for how well the mapped reads cover the viral genome. In general, coverage of greater than 0.5 indicates positve detection and coverage less than 0.2 indicates negative detection.
Depth: a measure of how many times a genome is covered by mapped reads.
Weight: the calculated proportion of reads mapping to a virus. The weight is roughly proportional to the titre. Higher the titre, higher the weight. A weight greater than 0.001 is a strong indicator of positive detection.
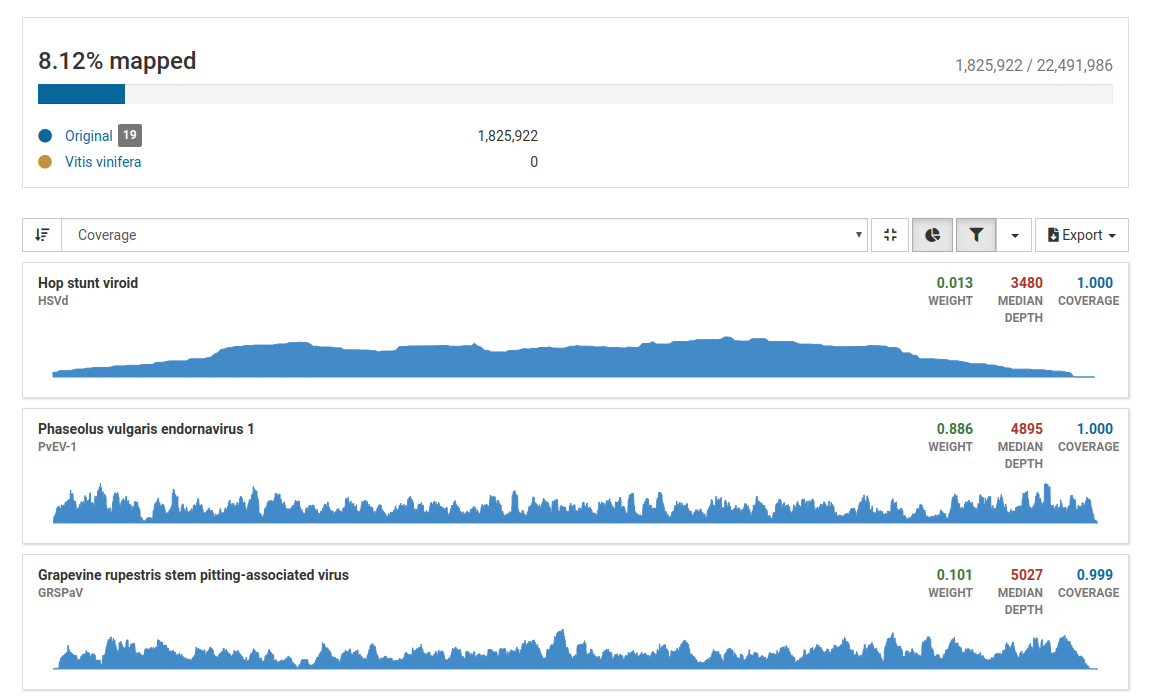
Three pathogens are likely to be in the sample. Although the total number of mapped reads is less than 5 million, the percentage of mapped reads is 8.12%, which is a good indicator of enrichment of viral RNA.
Additionally, all three pathogens have high weight, depth, and coverage values, therefore we can confidently say that indeed these pathogens are present in the sample.
The results from Pathoscope were successful in identifying the pathogens present in the sample, therefore further analysis using NuVs is not necessary. An indication that would result in using NuVs would be if our results showed high weight and depth values in combination with low coverage values. This would indicate that the virus sequence in the sample may be significantly different than what is in the database. This may represent a new genotype or possibly a new, closely related virus.